
Il Vera Prenatal Test® è un Test prenatale non invasivo che permette l’analisi del DNA fetale libero circolante, isolato da un campione di sangue materno, e valuta la presenza delle aneuploidie fetali comuni in gravidanza dei cromosomi 21, 18, 13 e dei cromosomi sessuali (X e Y), consentendo la determinazione del sesso fetale (opzionale).
Il fine del Vera Prenatal Test® è quello di fornire informazioni per le principali patologie riscontrate in gravidanza, alle gestanti/coppie che lo desiderano, affinché le successive scelte e decisioni, siano fondate su conoscenze il più possibile accurate, precoci e basate su protocolli che non mettono a rischio la gravidanza stessa. La consulenza prenatale è parte integrante dello screening mediante il Vera Prenatal Test® e ha lo scopo di aiutare la gestante/coppia nella comprensione del test, dei suoi benefici e dei suoi limiti nonché di definire un’eventuale alternativa al test stesso. Il Vera+ Plus, in aggiunta, prevede la possibilità, ove espressamente richiesto, di eseguire un approfondimento di secondo livello, per l’individuazione nel feto delle aneuploidie a carico di ogni cromosoma e delle alterazioni cromosomiche strutturali. Inoltre, grazie al “Pannello Microdelezioni”, è possibile valutare la presenza di alcune tra le più comuni microdelezioni, quali: la Sindrome di Di George, la Sindrome di Cri-du-chat, la Sindrome di Prader-Willi/ Angelman, la Sindrome da delezione 1p36, la Sindrome di Wolf-Hirschorn, la Sindrome di Jacobsen, la Sindrome di Langer-Giedion, la Sindrome di Smith-Magenis, la Sindrome da Brachidattilia-decit cognitivo.
In dettaglio:
Il Vera Prenatal Test®: rileva le aneuploidie a carico dei cromosomi 21, 18, 13 e dei cromosomi sessuali (X e Y) e consente anche la determinazione del sesso (opzionale).
Le aneuploidie sono anomalie cromosomiche caratterizzate da alterazioni del numero dei cromosomi, cioè da un numero maggiore o minore di cromosomi rispetto al numero standard. Il termine “trisomia” identica la presenza di tre, anziché di due, copie di un determinato cromosoma. Il termine “monosomia”, invece, identica l’assenza di una delle due copie di un cromosoma. Le anomalie numeriche dei cromosomi possono derivare tanto da alterazioni della meiosi durante la gametogenesi parentale, quanto da alterazioni mitotiche durante le prime fasi dello sviluppo dello zigote. Le più comuni forme di aneuploidia (trisomie e monosomie) derivano in genere da un processo di non-disgiunzione meiotica. Una più rara possibilità è quella della segregazione, per la quale uno dei due cromosomi gli viene ritardato in anafase, entrando a far parte della cellula nella quale è migratol’omologo. Se la non-disgiunzione mitotica si verica in una mitosi zigotica, ne deriva un mosaicismo, termine con il quale si indica una condizione caratterizzata dalla presenza nello stesso individuo di linee cellulari con due o più cariotipi diversi.
- La Trisomia 21 (T21) è l’aneuploidia più comune, e consiste nella presenza di una copia in più di un cromosoma 21 e si associa alla Sindrome di Down. Ha una prevalenza complessiva di un caso su 600-700 nascite. L’età materna è un fattore determinante la probabilità di nascita di un figlio Down: si passa da una probabilità di 1/2.500 prima dei 20 anni, a una di 1/700 tra i 30 e i 34 anni, 1/230 tra i 35 e i 39 anni, 1 /60 tra i 40 e i 44 anni, fino a una probabilità di circa 1/50 se l’età materna supera i 45 anni. Il fenotipo della sindrome di Down è caratterizzato da ritardo mentale associato, in genere, a carattere mansueto e socievole e da una serie di malformazioni. Le più tipiche comprendono: bassa statura, rima palpebrale obliqua (da ciò il vecchio termine, ormai obsoleto, di “mongolismo”), naso appiattito, bocca semiaperta con lingua grossa e solcata, spesso protrudente, orecchie prominenti con lobuli assenti, occipite piatto, collo corto e tozzo, mani tozze con mignoli arcuati, anomalie delle pliche palmari. Frequenti le malformazioni di organi interni, particolarmente a carico degli apparati cardiovascolare e renale. Vi può essere una deficienza della reattività immunologica. I soggetti affetti da sindrome di Down vanno più facilmente incontro a processi neoplastici e mostrano una incidenza di leucemia linfatica acuta pari a 20 volte maggiore di quella della popolazione generale. L’aspettativa media di vita di questi pazienti negli ultimi decenni è andata progressivamente allungandosi, circa 40 anni, grazie ai passi avanti nelle cure mediche. Le donne affette dalla sindrome di Down solo in rari casi sono feconde, i maschi costantemente sterili.

- La trisomia 18 (T18) consiste nella presenza di una copia in più di un cromosoma 18 e si associa alla Sindrome di Edwards. Presenta una frequenza di 1/6000-1/8000 nati, è più frequente nelle femmine ed è correlata con età materna avanzata. Nelle prime settimane di vita sono presenti ipotonia, iporeattività e difficoltà alimentari (scarsa suzione), seguite dalla progressione verso l’ipertono con l’apparente perdita della percezione dell’ambiente circostante. Le caratteristiche cliniche più comuni riguardano il ritardo della crescita prenatale e postnatale, l’aspetto emaciato con ipotroa, la microcefalia con cranio stretto e dolicocefalia, la microretrognazia, l’ipertelorismo, le orecchie angolate a disegno semplice. Le malformazioni sono comuni con interessamento oculare (microftalmia, coloboma), cardiaco (in oltre il 90% dei casi), del tubo digerente (atresia esofagea, malformazioni ano-rettali), del tratto genito-urinario (idronefrosi, agenesia mono-bilaterale). Il 90% dei bambini muore nel primo anno di vita a causa delle complicazioni cardiache, renali o neurologiche o delle infezioni ricorrenti.

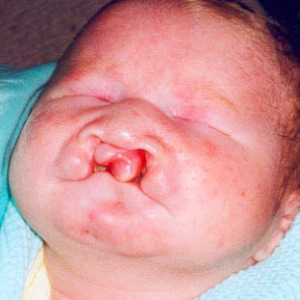
- La trisomia 13 (T13) consiste nella presenza di una copia in più di un cromosoma 13 e si associa alla Sindrome di Patau. La T13 ha una frequenza di 1/8000-1/15000 nati, e non è influenzata dall’età materna. Gli aspetti somatici sono rappresentati da grave ritardo psicomotorio, microftalmia (una malformazione che rende uno o entrambi gli occhi più piccoli del normale sebbene possano risultare strutturati in maniera normale), orecchie malformate, palatoschisi (malformazione congenita del palato), polidattilia (consiste in una malformazione in cui le dita della mano possono essere in eccesso), piede convesso (“a base di dondolo”); si associano malformazioni cardiache e renali. La trisomia 13 libera riguarda circa il 75% dei casi. Nel 20% dei casi, la trisomia 13 si associa alla traslocazione Robertsoniana nella quale il cromosoma soprannumerario 13 è attaccato a un altro cromosoma acrocentrico (cromosoma 13, 14, 15, 21 o 22). Nei feti affetti la morte endouterina si verifica in oltre il 95% dei casi. Rara la sopravvivenza oltre i primi mesi di vita.
Il Vera+ Plus®: consente di rilevare le aneuploidie a carico di tutti i cromosomi autosomici e le alterazioni cromosomiche strutturali del feto a carico di ogni cromosoma, quali duplicazioni e/o delezioni e/o traslocazioni sbilanciate per tutti i cromosomi con una risoluzione maggiore di 7 Mb.
Pannello Microdelezioni: Le Sindromi da Microdelezione sono anomalie caratterizzate dall’assenza di un tratto cromosomico di piccole dimensioni con conseguente perdita di informazione genica. Queste alterazioni causano sindromi di importanza clinica variabile a seconda del cromosoma coinvolto, della regione cromosomica interessata e delle relative dimensioni. Il Pannello Microdelezioni, aggiunto al Vera Prenatal Test® o al Vera+Plus®, valuta la presenza di alcune tra le principali sindromi da microdelezione, con una risoluzione ≥ 3,5 Mb. Le microdelezioni analizzabili sono quelle associate a: Sindrome di Di George, Sindrome di Cri-du-chat, Sindrome di Prader-Willi/Angelman, Sindrome da delezione 1p36, Sindrome di Wolf-Hirschorn, Sindrome di Jacobsen, Sindrome di Langer-Giedion, Sindrome di Smith-Magenis, Sindrome da Brachidattilia-deficit cognitivo.
- La Sindrome di DiGeorge associata alla delezione 22q11.2, è una delle più comuni sindromi da delezione cromosomica con un’incidenza mondiale di 1/2.000-1/4.000 nati vivi. Si manifesta con un quadro fenotipico estremamente variabile. Le più frequenti manifestazioni riguardano il sistema immunitario, con deficit immunitario da aplasia/ipoplasia del timo, che li rende suscettibili alle infezioni e le cardiopatie (77% dei casi) che riguardano soprattutto la regione troncoconale. Oltre il 75% dei pazienti presenta anomalie del palato (palatoschisi aperta, labiopalatoschisi, insufficienza velofaringea), che possono causare voce ipernasale, disturbi alimentari e della deglutizione. È frequente il ritardo dello sviluppo. Molti pazienti presentano lievi dismorsmi facciali (ipoplasia della regione malare, ptosi, ipertelorismo, epicanto, radice nasale prominente) e anomalie vertebrali (vertebre a farfalla, emivertebre);
- La sindrome di Cri-du-Chat associata alla delezione 5p15.3, è un disturbo genetico raro (incidenza di 1/50000) causato da una delezione del cromosoma 5 (5p-). L’ampiezza della delezione può variare molto, interessando la sola banda 5p15 o tutto il braccio corto del quinto cromosoma. La delezione può essere “de novo”, cioè verificarsi in maniera casuale, o essere il prodotto non bilanciato di una traslocazione bilanciata ereditata dal genitore sano. Le caratteristiche cliniche includono dismorsmi facciali, malformazioni viscerali, microcefalia, pianto dalla tonalità acuta e monotona simile al miagolio del gatto. La diagnosi viene fatta generalmente dopo la nascita e a causa della gravità della sindrome, da un punto di vista motorio e cognitivo, è importante intervenire precocemente entro i primi mesi di vita per operare in modo globale sullo sviluppo del bambino;
- La Sindrome di Prader-Willi/Angelman associata alla delezione della regione critica del cromosoma 15 è eterogenea dal punto di vista clinico e genetico. Approssimativamente nel 70% dei casi il difetto genico è dovuto a una delezione di 5-7 Mb del cromosoma 15 che include le bande 15q11.2-q13, nel 25-30% dei casi da disomia uniparentale materna del cromosoma 15 (udp(15)- mat), nel 1% dei casi a difetti di imprinting nella stessa regione del cromosoma 15 e in casi più rari da traslocazioni bilanciate con punto di rottura nei geni SNURF-SNRPN, o delezioni del gene SNORD 116. La malattia colpisce 1/25.000 nati ed è caratterizzata da anomalie ipotalamico-pituitarie associate a grave ipotonia nel periodo neonatale e nei primi due anni di vita, che comporta problemi alla deglutizione e all’allattamento. Dopo questa fase iniziale, i segni principali sono l’iperfagia e la mancanza di sazietà che causa spesso, nei bambini affetti di circa due anni, obesità grave. In assenza di controlli esterni adeguati, la condizione può peggiorare rapidamente, infatti è l’obesità la causa di mortalità più importante per i pazienti. Altre anomalie endocrine correlate contribuiscono a un quadro clinico caratterizzato da bassa statura, deficit dell’ormone della crescita, e sviluppo puberale incompleto. Il deficit cognitivo è estremamente variabile e si associa a difficoltà di apprendimento e a uno sviluppo anomalo del linguaggio, spesso aggravati dai disturbi comportamentali e psicologici;
- La Sindrome di Angelman è una malattia neurologica, di origine genetica, caratterizzata da grave ritardo mentale e dismorsmi facciali caratteristici. La sua prevalenza è stimata tra 1/10.000 e 1/20.000. I pazienti appaiono normali alla nascita. Nei primi 6 mesi di vita possono manifestarsi disturbi dell’alimentazione e ipotonia, seguiti da ritardo dello sviluppo tra i 6 mesi e i 2 anni. In genere, i sintomi caratteristici si manifestano a partire dal primo anno di vita, con grave ritardo mentale, assenza del linguaggio, crisi di riso associate a movimenti stereotipati delle mani, microcefalia, macrostomia, ipoplasia mascellare, prognatismo e disturbi neurologici con andatura da “burattino”, atassia e attacchi epilettici associati ad anomalie specifiche all’elettroencefalogramma (EEG; attività delta trifasica con picchi nelle regioni frontali). Altri segni clinici comprendono l’aspetto felice, l’iperattività senza aggressività, il basso livello di attenzione, l’eccitabilità, i disturbi del sonno associati a una riduzione della necessità di dormire, elevata sensibilità al calore e attrazione per l’acqua. Con l’avanzare dell’età, i segni tipici della malattia diventano meno accentuati. La malattia è causata da diversi meccanismi genetici, come la delezione nella regione critica 15q11.2-q13 (60-75% dei casi), la disomia uniparentale paterna (2-5%), un difetto dell’imprinting (2-5%) e la mutazione del gene UBE3A (10%). Nel 5-26% dei pazienti non è stato identificato il difetto genetico.
- La Sindrome da delezione 1p36 è causata da una delezione parziale della parte distale del braccio corto del cromosoma 1. Circa il 50% dei casi è dovuto a una delezione “de novo” 1p36 terminale, circa il 29% a una delezione interstiziale; gli altri casi riguardano riarrangiamenti cromosomici più complessi. È considerata una delle più comuni sindromi da delezione cromosomica, con un’incidenza di 1/5.000-10.000 nati vivi. Colpisce in uguale misura entrambi i sessi, in tutte le etnie. I pazienti presentano caratteristici dismorfismi craniofacciali con sopracciglia diritte, occhi infossati, ponte/sella nasale larga e piatta, ipoplasia mediofacciale, ltro lungo, mento appuntito e, spesso, fontanella anteriore gran-de con chiusura tardiva (>3 cm alla nascita), microbrachicefalia, orecchie anomale, ruotate posteriormente, a basso impianto. Sono anche caratteristici la brachidattilia, la campodattilia e i piedi corti. Quasi tutti i pazienti presentano ipotonia congenita, che concorre alle di‑coltà all’alimentazione, al ritardo dello sviluppo motorio e della motilità, al ritardo o all’assenza del linguaggio. In tutti i pazienti è presente ritardo mentale variabile.
- La Sindrome di Wolf-Hirschorn è dovuta a una delezione del braccio corto del cromosoma 4, che comprende almeno parte dei geni LETM1 e WHSC1. La prevalenza è di 1:50.000 nati. Interessa più spesso le femmine rispetto ai maschi (2:1). Si osservano marcato ritardo della crescita prenatale, lentezza costante nel guadagno del peso postnatale, una facies tipica ad “elmo da guerriero greco” (radice del naso larga che continua sulla fronte) molto più evidente prima della pubertà, microcefalia, fronte alta con glabella prominente, ipertelorismo, epicanto, sopracciglia molto arcuate, filtro corto, bocca rivolta verso il basso, micrognatia, orecchie poco formate con fossette/appendici e, in alcuni casi, labiopalatoschisi. Sono presenti anomalie scheletriche, come la cifosi o scoliosi con malformazione dei corpi vertebrali, costole fuse o accessorie, piedi torti e schisi delle mani. È presente ipotonia con ipoplasia delle masse muscolari, che può associarsi a difficoltà alimentari e contribuire al ritardo della crescita. Il ritardo dello sviluppo è grave: molti pazienti non imparano a controllare lo sfintere, a mangiare e vestirsi autonomamente, e meno del 50% cammina, con o senza sostegni, tra i 2 e i 12 anni di età. Il deficit cognitivo è moderato grave, di rado lieve. Il linguaggio si limita a suoni gutturali o bisillabici e, in alcuni casi, a semplici frasi.
- La Sindrome di Jacobsen associata alla delezione del (11)(q23), è una malattia causata dalla delezione parziale del braccio lungo del cromosoma 11, caratterizzata da anomalie congenite multiple/ritardo mentale (MCA/MR), la prevalenza è stimata in circa 1/100.000 nati, con un rapporto femmine/maschi di 2:1. I segni clinici più comuni sono il ritardo di crescita pre- e post-natale, il ritardo psicomotorio e i dismorsmi facciali (deformità del cranio, ipertelorismo, ptosi, coloboma, rime palpebrali rivolte verso il basso, epicanto, sella nasale ampia, naso corto, labbra a forma di V, orecchie piccole, a bassa attaccatura e retroruotate). Le dimensioni della delezione variano da ~7 a 20 Mb, con punto di rottura prossimale nella sottobanda 11q23.3 o in una regione più telomerica. La delezione si estende di solito fino al telomero. La delezione è “de novo” nell’85% dei casi, mentre nel 15% deriva dalla segregazione sbilanciata di una traslocazione familiare bilanciata o da riarrangiamenti che coinvolgono altri cromosomi. Circa il 20% dei bambini muore nei primi due anni di vita, più spesso per le complicazioni della cardiopatia e, meno frequentemente, per le emorragie. Nei pazienti che superano il periodo neonatale e l’infanzia, le attese di vita non sono note.
- La Sindrome di Langer-Giedion, associata alla delezione 8q24.11-q24.13, è caratterizzata da deficit cognitivo, associato a varie anomalie, compresa la cute ridondante, le esostosi cartilaginee multiple, la facies caratteristica e le epifisi falangeali “a cono”. La gravità e il numero di queste anomalie variano nei diversi pazienti. I dismorfismi facciali comprendono il naso globoso, il filtro ampio e prominente, il labbro superiore sottile, le orecchie “a cavolfiore”, i capelli radi e l’ipoplasia mandibolare. Sono stati descritti anche ritardo di crescita, microcefalia, ipotonia e problemi uditivi. Le esostosi (tumori ossei benigni) si localizzano prevalentemente sulle estremità delle ossa lunghe e possono causare dolore, rigidità funzionale o deformazione. Si trasmette come carattere autosomico dominante, ma sono stati descritti soprattutto casi sporadici. La precocità della diagnosi è fondamentale, ai fini della consulenza genetica alle famiglie e per garantire il follow-up ortopedico e la gestione dei problemi della crescita e dell’udito.
- La Sindrome di Smith Magenis, associata alla delezione 17p11.2, è causa della delezione del gene RAI1 nel 90% dei casi (un altro 10% è associata a mutazioni nello stesso gene). La prevalenza mondiale è di 1/15.000-25.000 in tutti i gruppi etnici, ma è probabile che sia sottodiagnosticata. I segni clinici comprendono segni craniofacciali (brachicefalia, fronte bombata, ipertelorismo, sinofria, rime palpebrali oblique verso l’alto, ipoplasia mediofacciale, faccia quadrata larga con sella nasale depressa, labbro superiore rovesciato a “tenda”, micrognatia neonatale), altre anomalie scheletriche (brachidattilia, scoliosi, clinodattilia del V dito delle mani, sindattilia delle dita dei piedi 2-3, movimenti limitati dell’avambraccio e del gomito, anomalie vertebrali, persistenza dei rigonfiamenti fetali sui polpastrelli delle dita delle mani, polidattilia), frequenti disturbi otorinolaringoiatrici, segni oftalmologici (>60: miopia, anomalie iridee, di rado distacco della retina spesso in seguito a comportamenti violenti). La perdita dell’udito (60% dei pazienti) è variabile e può essere lieve-moderata. Sono comuni il deficit cognitivo lieve-moderato, il ritardo significativo del linguaggio, la ridotta sensibilità al dolore, la neuropatia periferica, i disturbi del sonno (caratteristici) e i comportamenti disadattivi (capricci/scatti d’ira, ricerca costante dell’attenzione, aggressività, disobbedienza, distrazione e comportamenti autolesionistici). Le malformazioni a livello degli organi (30-40%) sono le cardiopatie, le anomalie renali, urinarie e del SNC. La delezione 2q37, brachidattilia-deficit cognitivo, è un’anomalia cromosomica con delezione della banda cromosomica 2q37 e si manifesta con tre sintomi principali: ritardo dello sviluppo, malformazioni scheletriche e dimorfismi facciali. L’incidenza è stimata in meno di 1 ogni 10.000. La maggior parte dei pazienti presenta ritardo dello sviluppo o deficit cognitivo, bassa statura (23% dei casi), tendenza all’obesità con l’avanzare dell’età e brachimetafalangia (50%). Sono anche frequenti la clinodattilia del quinto dito e la sindattilia delle dita delle mani e dei piedi, la microcefalia o la macrocefalia. I dimorfismi facciali sono caratteristici e comprendono il viso arrotondato, i capelli radi, la fronte prominente, le rime palpebrali oblique verso l’alto, gli occhi infossati, le sopracciglia rade e arcuate, l’ipoplasia della porzione media del viso, la radice nasale infossata, le anomalie delle ali del naso, la columella prominente, l’aspetto a V della punta del naso, l’assottigliamento delle labbra e il palato ogivale. Nel 30% dei pazienti sono presenti malformazioni, in particolare cardiopatie, difetti gastrointestinali (30%), urogenitali (11%) e del sistema nervoso centrale (6%). È spesso presente ipotonia. Sono state riportate convulsioni in circa il 35% dei pazienti e anomalie del comportamento in circa il 30%.
Modalità di esecuzione:
Durante la gravidanza, alcuni frammenti del DNA del feto (cDNA) circolano nel sangue materno. Tale DNA proviene dal citotrofoblasto (un tessuto che fornisce nutrimento all’embrione) che forma la placenta. Il ricambio delle cellule del trofoblasto, che ricopre le pareti delle arterie spiraliformi, mediato dalle citochine, libera il DNA. Diversi studi hanno dimostrato che il DNA fetale può essere trovato già alla 7ª settimana di gestazione e la sua quantità aumenta con il progredire della gravidanza e scompare subito dopo il parto. La percentuale di DNA fetale può variare tra <4%, una quantità non utile per la diagnosi, e il 40%, con una media del 10%, alla 12ª settimana. La percentuale del cffDNA presente nel plasma materno viene definita “frazione fetale” (FF). La quantità di DNA fetale circolante dalla 10ª settimana di gestazione è sufficiente per garantire l’elevata specificità e sensibilità del test. Presso il Centro Diagnostico Polispecialistico Strumentale Ames di Casalnuovo è possibile eseguire il test prenatale non invasivo Vera Prenatal Test® con marcatura CE-IVD, che combina la tecnologia di sequenziamento di nuova generazione (NGS) Illumina con la postazione automatizzata VeriSeq NIPT Microlab® STAR™ della Hamilton Robotics fornendo un sistema di sequenziamento per l’intero genoma con il più basso failure rate (tasso di fallimento) tra le tecnologie utilizzate per le NIPT. Durante la fase di sequenziamento massivo entrambi i lamenti del DNA sono analizzati, permettendo così di determinare la grandezza di ogni frammento di DNA libero (cffDNA) nel campione, infatti nel sangue materno è contenuto DNA libero di differente lunghezza: i frammenti cffDNA più corti sono di origine fetale mentre i frammenti più lunghi sono materni. Il sistema di sequenziamento usato è in grado di migliorare il rapporto segnale/rumore di fondo selezionando frammenti più corti (fetali) ed effettuando letture molto accurate del frammento utilizzando meno di un terzo della profondità di lettura di altre metodiche di sequenziamento. Questo consente di risparmiare tempo e risorse rispetto ai protocolli di lettura singola, producendo risultati NIPT in modo rapido e conveniente. L’automatizzazione del test consente di ridurre i tempi e i potenziali errori introdotti nella fase analitica. Partendo da 7-10 ml di sangue materno, il VeriSeq NIPT MicrolabSTAR consente di controllare tutti i passaggi della preparazione del campione, inclusi la separazione del plasma, l’estrazione del DNA libero e la preparazione della libreria. Le librerie preparate dai campioni vengono sequenziate con strumenti IlluminaNextSeqTM 500, NextSeqTM 550 di cui è dotato l’AMES. Una volta completato il sequenziamento, i dati vengono inviati automaticamente al VeriSeq NIPT Assay Software per l’analisi e la generazione dei report. Il test VeriSeq NIPT Solution utilizza una procedura che non prevede l’utilizzo della PCR che potrebbe eventualmente introdurre alterazioni nella lettura, fornendo così una visione più completa del materiale genomico. Inoltre, avendo dati di copertura disponibili per l’intero genoma diploide, si produce un riferimento analitico che le attuali tecniche analitiche possono utilizzare per ridurre bias legati alla tecnica e al campione. Questi passaggi di normalizzazione si traducono in un failure rate (percentuale di fallimenti, cioè l’assenza di risultati per disomia o aneuplodia) più basso rispetto agli altri test di sequenziamento che utilizzano un approccio “targeted”, riducendo così la necessità di attuare interventi invasivi, quali l’amniocentesi, per avere informazioni sulla salute del feto.
!!ATTENZIONE!!
I test non sostituiscono la diagnosi prenatale invasiva (Amniocentesi o Villocentesi)
Lo screening prenatale non invasivo basato sul DNA non è un test diagnostico. Il test verifica la possibilità che il feto sia affetto dalle più comuni aneuploidie, con una specificità e sensibilità significativamente superiori rispetto allo screening non invasivo combinato (TN+PAPP-A/βHCG). Il test potrebbe dare un risultato positivo in caso di mosaicismo cromosomico, ma questo potrebbe essere connato alla placenta. Sebbene il test definisca, la presenza nel feto di una specifica patologia indagata, resta un test di screening, pertanto, ogni risultato positivo deve essere confermato con una tecnica invasiva tradizionale (villocentesi/amniocentesi). Il test è stato validato su gravidanze singole o gemellari, monozigotiche o dizigotiche, con almeno 10 settimane di gestazione. L’esame infatti non è in grado di evidenziare riarrangiamenti cromosomici bilanciati, alterazioni cromosomiche strutturali, alterazioni parziali dei cromosomi analizzati, mosaicismi cromosomici fetali e/o placentari (cioè la presenza di due linee cellulari con differente assetto cromosomico), mutazioni puntiformi, delezioni/duplicazioni di uno o più esoni, difetti di metilazione, poliploidie. Il test non evidenzia altre malformazioni o difetti non specificamente ricercati. In particolare, l’esame non evidenzia la presenza di malattie genetiche ereditarie a trasmissione mendeliana. Le gravidanze con riscontri ecograci suggestivi di patologia fetale dovrebbero essere studiate con altri tipi di indagini prenatali, quali il cariotipo fetale molecolare su villi coriali o liquido amniotico, in considerazione del maggiore detection rate. Nelle gravidanze gemellari non è possibile distinguere la condizione del singolo feto, né di valutare le aneuploidie dei cromosomi sessuali, ma solo quelle relative ai cromosomi 13, 18, 21. E’, tuttavia, possibile riscontrare la presenza/assenza del cromosoma Y. Nel caso in cui venga individuata la presenza del cromosoma Y, non è possibile valutare se solo uno o entrambi i feti siano di sesso maschile. Nelle gravidanze iniziate come gemellari o plurime, seguite dall’aborto spontaneo di uno o più feti con riassorbimento della camera gestazionale, potrebbe essere presente nel sangue materno anche il DNA fetale libero del feto abortito. Ciò potrebbe interferire nella qualità dei risultati, determinando falsi positivi nel caso in cui la causa dell’aborto fosse dovuta alla presenza nel suddetto feto di aneuploidie cromosomiche a carico di uno dei cromosomi investigati. Similmente, potrebbe determinarsi una incongruenza nei risultati del sesso (es. diagnosi di sesso maschile, in cui la presenza del cromosoma Y è originata dal DNA feto abortito). Non è possibile eseguire il test a gravidanze plurigemellari (trigemellari o più).
Per info riguardanti il Vera Prenatal-Test® contattare i numeri:
- Fisso: 0761347138
- Mobile: 392/9594263
- WhatsApp: 392/9594263
Argomenti correlati
Servizi correlati
- Screening Prenatale Non Invasivo Molecolare (NIPT)
- Screening Genetici Preconcezionali della Coppia
- Screening Prenatale Non Invasivo-Genetica Biochimica
- Analisi Citogenetica Classica e Molecolare
- Onocologia Molecolare – Test Predittivi (Pannelli NGS)
- Oncologia Molecolare (Diagnosi Precoce)
- Genetica Cardiovascolare
- Osteoporosi
